私たちはSR11000を使って研究しています ー高速演算サーバSR11000利用の成果ー
SR11000 を用いたMPI並列計算による境界要素法の加速
(平成17年度当センター公募プロジェクト研究)
宮川悠(自然科学研究科科・博士後期課程)
植田毅(当センター 大規模情報システム研究部門 助教授)
1.MPI並列計算
近年,半導体技術の発展により計算機の能力が飛躍的に発達し,それに伴い今まで不可能だと思われていたさまざまな科学技術計算が可能となってきた。しかし,大規模計算と呼ばれる分野では一つのCPU ではまだまだ計算能力が不足しており,多数のCPUを用いた並列計算による加速が盛んに行われている。そこで本研究では,千葉大学総合メディア基盤センターに設置されているSR11000を用いてMPI並列計算により境界要素法を加速させる。現在の並列計算機は大きく分けて2 つあり,全てのプロセスが同じメモリーを共有する共有メモリー型(SMP並列計算) とそれぞれのプロセスが異なるメモリーを持つ分散メモリー型(MPI 並列計算) がある。 SR11000はこの2つの特徴を持つSMP クラスター型と呼ばれている。図1 は,量子細線中に点状散乱体を三角格子状に配置し,点状散乱体を扱えるよう独自に拡張した境界要素法により電子の確率密度を計算したものである。本研究ではこの境界要素法をMPI 並列計算を用いて加速を行う。

2.分割法の決定
多くの並列計算ではループの分割により並列化を行う。もし理想的な分割を行えば一つのループが終わるまでの計算時間を1/CPU 数とすることができる。このループの分割法はいくつか考えられる。まず,ループを単純に全プロセス数で割ったブロック分割では,要素数は波数kdに比例して大きくなるため,波数が低い部分と高い部分とでロードバランスが不均等になるため効率がよくない。またSR11000は全て同じCPUで構成されており,それぞれのノードでの処理能力にばらつきがないので,要素数が近ければ計算時間もほぼ同じだと考えられる。それをふまえて,それぞれのプロセスが総プロセス数ごとに繰り返すサイクリック方式を採用する事とした。
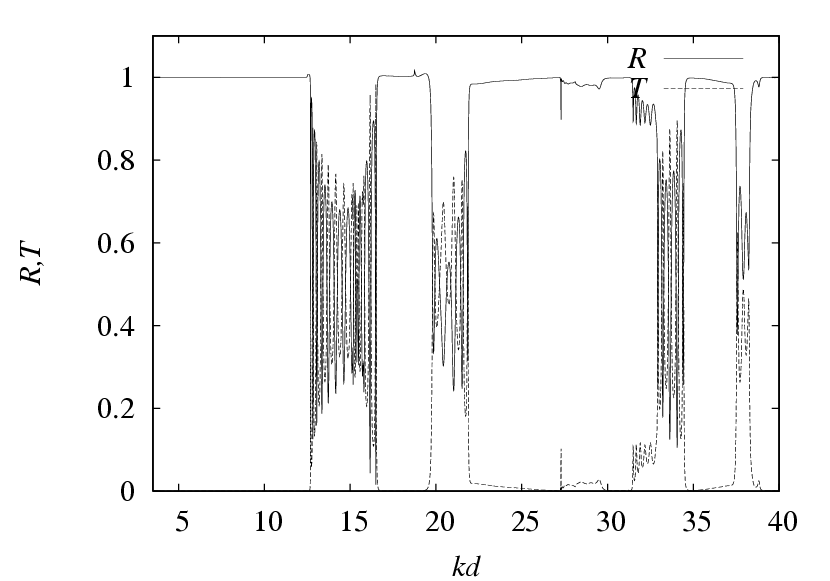
3.計算時間
図2は,図1のような不純物がある場合を境界要素法により透過,反射スペクトルを計算したものである。この計算をSR11000で1CPUで行ったときの計算時間は16時間,対してMPI並列計算により2ノード,32CPUを用いて行ったときの計算時間は30分であった。ほぼCPU数倍の加速が実現していることが分かる。このようにSR11000によるMPI 並列計算を用いれば,境界要素法によるスペクトルの計算を,現実的な計算時間で行うことが可能となった。
原子核の新しい平均場描像(投稿)
中田仁(理学部 物理学科 助教授)
1980年代半ばに不安定核ビームの実験技術が開発されて以降,不安定核の構造が理論・実験の両面において原子核物理学の大きなtopicになっている。 2007年からは理化学研究所において次世代の不安定核ビームが出始め,日本が世界をリードする形で実験的研究が進む。ところで,核内で核子(陽子と中性子の総称)は概ね「1粒子軌道」上を運動している。しかし今までの研究により,不安定核ではこの核子軌道の性質が安定核の場合とかなり異なることが分かってきた。これを理解するには,1)核子の束縛の程度(典型的には束縛エネルギー)による軌道の性質の違いや,2)核子間の有効相互作用の性質,を再検討する必要があり,3)そのための数値計算法の開発も望まれる。我々は,この3点にweightをおいて研究を進めている。
核内核子の軌道を自己無撞着に求めるため, Hartree-Fock(HF)理論やHartree-Fock-Bogolyubov(HFB)理論が用いられる。これらの平均場理論を原子核に適用する際,従来は主として実用上の理由から相互作用の形を強く限定していた。その制限を取り除き,かつ上記1)の点にも対応できる方法として,我々は「ガウス関数展開法」を採用し,その有効性を検証している[1,2]。ガウス関数展開法はもともと少数系の精密数値計算のために発案され応用されてきた[3]が,そのアイディアを生かし平均場計算に適合するアルゴリズムを開発したのである。最近の文献[2]では,複素レンジのガウス関数を用いてHFB理論による数値計算が有効に行えることを,本学総合メディア基盤センターのHITACHI SR11000を利用して具体的に数値計算を実行することにより示した。この方法が様々な相互作用に対する高い適合性を持つことは,方法の構成上明らかである。今後,アルゴリズムの拡張として回転対称性の破れたより複雑な系への適用を行いたい。またより広い視点からは,多数の原子核での数値計算を通じて,相互作用の再検討も含めた総合的な不安定核の研究へと発展させたいと考えている。
[1] H. Nakada and M. Sato, Nucl. Phys. A 699, 511 (2002); ibid 714, 696 (2003).
[2] H. Nakada, Nucl. Phys. A 764, 117 (2006).
[3] 肥山, 木野, 上村, 日本物理学会誌 61, 27 (2006)
標的タンパク質とリガンドの結合様式予測プログラムの開発
(平成17年度当センター公募一般研究)
藤 秀義(医学薬学府・修士課程)
片桐大輔(薬学部・博士課程)
星野忠次(薬学研究院助教授)
研究目的
新薬開発におけるStructure -Based Drug Design (SBDD)の重要性は増している。 SBDDとは,タンパク質の立体構造情報に基づいて薬剤を設計する方法である。しかしながら多くの場合,標的とするタンパク質は,作用を受ける薬物や生理活性物質など(リガンドと呼ばれる)の種類とその結合部位が未知である。従って,結合部位をコンピューターで正確に決定する技術の確立は,SBDDの成功に不可欠である。本研究では,リガンドの結合部位,結合様式,親和性を評価するプログラムを開発する。
研究方法
Protein Data Bank(PDB: http://www.pdb.org/pdb/)[1]に登録されたタンパク質結晶構造に対して,タンパク質周囲に一定間隔で格子点を発生させ,各格子点における疎水性ポテンシャルを計算した。使用した疎水性ポテンシャルの計算式を式(1)に示す。
ΔGH = - 2.0R exp(-D/10) ・・・ (1)
ここで,ΔGHは疎水性エネルギー(kcal/mol). R = R1R2/(R1 + R2), R1はタンパク質中の炭素原子の半径, R2は格子点上のプローブ炭素原子の半径である。 D = R12 - (R1 - R2), R12はタンパク質中の炭素原子と格子点間の距離である。計算には,タンパク質中の疎水性残基(Gly, Ala, Val, Leu, Ile, Met, Trp, Phe, Pro)のカルボニル炭素を除く炭素原子を用いた[2]。各格子点における疎水性エネルギーを計算後,エネルギーの低い方から格子点100個を取り出し,格子点の多く集まった領域を,リガンド結合部位とした。
研究結果
立体構造が既知である25種のタンパク質-リガンド複合体について,リガンド結合部位の予測を行った。格子点を1Å間隔で発生させて計算したところ,15種のタンパク質のリガンド結合部位の同定に成功した。成功例のいくつかを図1に示す。
|
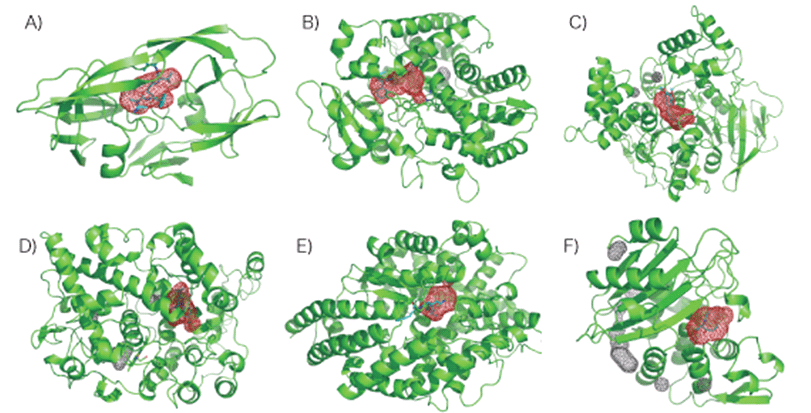
図1: 計算で同定されたリガンド結合部位(格子点間隔1Å).リガンドは水色のスティック表示で示し,疎水性領域はメッシュ表示で示した。疎水性領域のうちリガンド結合部位と予測した部位を赤で示した。A) HIV-1 protease (PDB ID:1AAQ), B) CYP2C9 (PDB ID: 1OG5), C) Acetylcholinesterase (PDB ID: 1EVE), D) Cyclooxygenase-2 (PDB ID: 6COX), E) Angiotensin converting enzyme (PDB ID: 1O86), F) β-lactamase (PDB ID: 1BLC)。
|
図1を見れば明らかなように,リガンド結合部位と疎水性領域の集まった部分が一致していることが分かる。次に,先程の方法で予測の出来なかった10種のタンパク質-リガンド複合体について,格子点を0.5Å間隔で発生させて計算した。すると,3種のタンパク質のリガンド結合部位の同定に成功した。その結果を図2に示す。
|
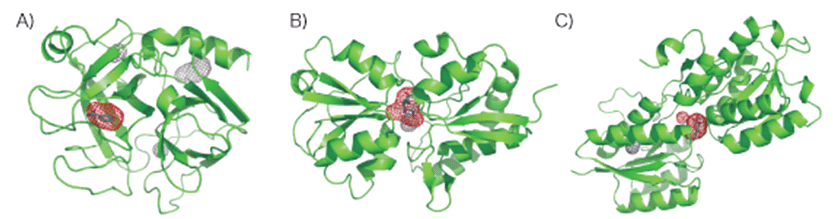
図2: 計算で同定されたリガンド結合部位(格子点間隔0.5Å)。表示の仕方は図1と同様。A) β-trypsin (PDB ID: 3PTB), B) Histidine-binding protein (PDB ID: 1HSL), C) D-galactose/D-glucose binding protein (PDB ID: 2GBP)。
|
考察
格子点間隔1Åで計算したところ,25種類中15種についてタンパク質のリガンド結合部位の同定に成功した。格子点間隔0.5Åで計算すると,25種類中18種について同定することができ,およそ70%程度の精度でリガンド結合部位の予測ができることがわかった。格子点間隔を細かくすることで予測に成功したタンパク質-リガンド複合体3種(図2)に着目してみると,結合しているリガンド分子が小さいということが共通して言える。 3PTB, 1HSL, 2GBPのリガンドの分子量は,それぞれ121.16, 155.16, 180.15であり,リガンドの溶媒露出表面積を見ても,312.58Å2, 334.12Å2, 341.09Å2と小さな分子であることが分かる(Ligand-Protein Database (LPDB)より[3])。従って,リガンド分子の大きさによって格子点間隔を変えて計算する必要があると考えられる。予測の出来なかったものには,ヘテロ2量体のタンパク質や,リガンド分子の親水性が高いものが含まれる。これらの複合体に対しては,計算方法の改良の余地がある。
今回,千葉大学総合メディア基盤センターの並列型スーパーコンピュータSR11000を使用することによって,Alpha21164 CPU(533MHz)搭載機で計算した時と比べて最大約70倍の計算速度の向上を図ることができた。本研究にて作成したリガンド結合部位予測プログラムを様々なCPUで動作させ,計算時間を比較した図を図3に示す。一般的に良く使われるAMD系やIntel系のCPUで動作させた時と比較しても,10数倍の高速化に成功している。このように,SR11000を用いることによって大幅な計算時間の削減を行なうことができる。よって,大量の化合物ライブラリーを扱った場合にも,迅速かつ効率良く有効な薬剤を見つけ出すことが可能になると期待される。
参考文献
[1] H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, P. E. Bourne, Nucleic Acids Res 28, 235-42 (2000).
[2] N. Yamaotsu, S. Hirono, The 3rd Annual Meeting of Chem-Bio Informatics Society 140-1 (2002).
[3] O. Roche, R. Kiyama, C. L. Brooks, J Med Chem 44, 3592-8 (2001).
|
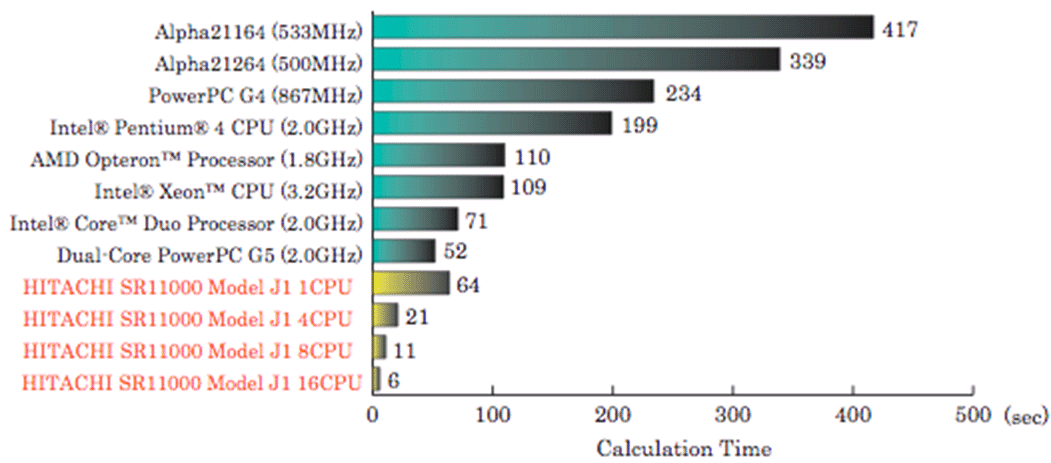
図3. リガンド結合部位予測プログラムの各CPUにおける計算速度
|
高速演算サーバご利用にあたっては,下記のURLをご覧下さい。
http://www.imit.chiba-u.jp/service/ed_rsch/guide.html
|